
Unappreciated lncRNAs, identification and spatial relationship to coding genes
Vikram R. Paralkar et. al have shown that lncRNAs are co-regulated with coding genes and can influence the maturation of erythroblasts. Erythroblasts are lineage from HSPCS (CD 34, 45, 59) which produce erythrocytes (red blood cells). LncRNAs are non-coding RNAs with at least 200 nucleotides in length. Paralkar et.al used polyA+ selection and deep sequencing to address the hypotheses 1) Do erythroid progenitors express lineage specific lncRNAs? 2) how are lncRNAs regulated?
Their manuscript developed an algorithm for identifying novel, unannotated, lncRNAs using 3 different algorithms which identify the non-coding potential of targeted regions. The algorithms used were 1) BlastX and Pfam protein database homology 2) codon conservation using PhyloCSF 3) presence of long open reading frames > 100 amino acids using EMBOSS GetORF.
Algorithm 1) can determine the absence of protein creation, hence non-coding and 2) 3) can assess the long non-coding potential by studying the homology and open-reading frames. Using the 3 algorithms they can cross-validate which potential novel lncRNAs are high-strigent and low strigent classifications. The HS-lncRNA classification had 64% novel lncRNAs that were not identified in RefSeq, UCSC, and ENSEMBL which is quite impressive because ENSEMBL offers leading annotation updates.
Comparing the lineage specificity, 90% coding gene mRNAs were found in all three clone types MEPs, Megakaryocytes, erythrocytes, whereas only 33% of the high-stringency lncRNAs were found in all three types. This indicates that lncRNAs may be lineage specific. Comparing lncRNAs to coding TSS, the proportion of HS/LS lncRNAs were equally frequent in diverge, antisense, intronic, and intergenic regions. In comparison to coding genes the orientation of lncRNAs is uniform, without specification of spatial relationships (ORF) to coding genes.
Transcription factors regulating mouse eyrthro-megakaryocytic lncRNAs
This group investigated promoter and enhancer signatures to determine how lncRNAs are transcribed. They used Chip-Seq for H3K4me1, H3K4me3, H3K436me3 histone marks on liver erythroblasts and megakaryocytes and mapped the chromatin signatures to genomic loci of coding genes and lncRNAs. Constructing a ratio defining promoters as H3K4me3-high/H3K4me1-low, and defining enhancers as H3K4me1-high/H3K4me3-low. ~25% of the TSS of lncRNAs had enhancer-like signatures which was significantly greater than coding genes (<10%) (Fisher’s Exact Test p=

To investigate the TF regulation, they performed Chip-Seq for GATA1, and TAL1 (erythroblasts) and GATA1,GATA2, TAL1, FLI1 in megakaryocytes (ENCODE definitions of lineage specific transcription factors). Vikram et. al compared binding of TFs in all three cell types (lncRNAs and coding genes) and found that the TFs were present uniformly frequently. In erythropoiesis, GATA1 was downregulated showed significantly higher frequencies in HS lncRNAs. The meaning of this is unclear, but implies shared regulation because of the uniform presence of known TFs.
Vikram then tested experimentally (in-vitro) the role of GATA1 regulation of erythroid lncRNAs. They used an immortalized Gata1
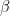
Interestingly, Vikram et. al have recently demonstrating the heptad TF (Gata2, Runx1, Tal1, Fli1, Lmo2, Ldb1, Lyl1, and Erg) in HSPCs were lineage specific activation with respect to coding genes were upregulated during megakaryopoiesis and repressed during erythropoiesis. To verify this regulation pattern in lncRNAs, they assessed the heptad TF occupancy and found the highest frequency occupancy was up activation in megakaryopoiesis and either down activation in erythropoiesis or no change in erythropoiesis. They showed that the TF heptad previously explored in coding genes were also primed in HSPCs as a common regulatory TF in lncRNAs under specific lineages.
RNAi to assess erythroid lncRNA function
They selected abundant and erythroid-enriched lncRNAs both conserved across mouse and human and non-conserved. They selected candidate lncRNAs that were significantly expressed in erythroblasts compared to other MEPs and excluded LS-lncRNAs. They also performed enrichment testing and confirmed the candidates were signficantly enriched in adult bone marrow erythroblasts using microarray analysis. Then they created shRNAs and selected 21 lncRNAs with at least 40% knockdown of lncRNA expression. Next they assessed the maturation of erythroblasts from lncRNA-knockdown erythroblasts and identified 1 LS-lncRNA and 6 HS-lncRNA whose knockdown by at least to different shRNAs. Of the novel 7, one was previously annotated “Lincred1”, and they’ve named the other 6 (Galont, Redrum, Erytha, Scarletltr, Bloodlinc, Ggnbp2). The knockdown affect on erythroid maturation implies that these 7 lncRNAs are involved with terminal maturation of erythroid cells.
Interestingly 6 out of 7 of the lncRNAs were conserved, yet showed no transcript expression in humans implying that interspecies lncRNAs expression does not guarantee functionality.
Vikram et. al 2014.
 which assumes no variants exist at the site and any non-reference target is due to sequencing error. The comparison group is defined as
which assumes no variants exist at the site and any non-reference target is due to sequencing error. The comparison group is defined as  where f is the estimated allelic fraction and m is the unknown true mutant at a site. f is estimated given the following:
where f is the estimated allelic fraction and m is the unknown true mutant at a site. f is estimated given the following: ,
,  is the base of the read and d is the site and
is the base of the read and d is the site and  is the probability of error of that base call denoted
is the probability of error of that base call denoted  where
where  is the Phred quality score. Given the Reference/comparison:
is the Phred quality score. Given the Reference/comparison:



 is piecewise set by the empirical observations of the mutant site matching or mismatching the reference. If the reference is matched then the allelic frequencing is penalized by a factor of the sequencing error and summed with the product of the complement of allele frequency and sequencing accuracy. If the base site matches a mutant then the allele frequency is multiplied by the sequencing accuracy and the complement allele frequency is then penalized. Normally the allele frequency is a small number so if a base matches the reference we expect the allele gene variant to be penalized and the non-gene variant frequency to be rewarded. If the base does match the mutant then the allele gene variant is expected to be rewarded and the non-gene allele variant to be penalized uniformly for all substitutions.
is piecewise set by the empirical observations of the mutant site matching or mismatching the reference. If the reference is matched then the allelic frequencing is penalized by a factor of the sequencing error and summed with the product of the complement of allele frequency and sequencing accuracy. If the base site matches a mutant then the allele frequency is multiplied by the sequencing accuracy and the complement allele frequency is then penalized. Normally the allele frequency is a small number so if a base matches the reference we expect the allele gene variant to be penalized and the non-gene variant frequency to be rewarded. If the base does match the mutant then the allele gene variant is expected to be rewarded and the non-gene allele variant to be penalized uniformly for all substitutions.
 ,
,  we assume them independent and
we assume them independent and  is uniformly distributed and in practive
is uniformly distributed and in practive  due to a reasonable assumption that typical mutational frequency is
due to a reasonable assumption that typical mutational frequency is  which is odds ratio of 10:1 in favor of the reference model which is to say that 1-10 mutational variant per 1Mb yielding
which is odds ratio of 10:1 in favor of the reference model which is to say that 1-10 mutational variant per 1Mb yielding  across all 3 mutational values of m allows the estimation of
across all 3 mutational values of m allows the estimation of 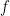 using maximum liklihood estimation to find
using maximum liklihood estimation to find 

